
Imperial College Computational Bioinformatics Laboratory (CBL)
Research
This page contains:-
- an overview of scientific knowledge discovery tasks carried out using Inductive Logic Programming (ILP);
- a bibliography of the corresponding publications;
- links to other pages on current and proposed projects.
Scientific Knowledge Discovery using ILP
The pharmaceutical industry is increasingly overwhelmed by large-volume-data. This is generated both internally as a side-effect of screening tests and combinatorial chemistry, as well as externally from sources such as the human genome project. On the other hand the industry is predominantly knowledge-driven. For instance, knowledge is required within computational chemistry for pharmacophore identification, as well as for determining biological function using sequence analysis.
From a computer science point of view, the knowledge requirements within the industry give higher emphasis to ``knowing that'' (declarative or descriptive knowledge) rather than ``knowing how'' (procedural or prescriptive knowledge). Mathematical logic has always been the preferred representation for declarative knowledge and thus knowledge discovery techniques are required which generate logical formulae from data. Inductive Logic Programming (ILP) [8,1] provides such an approach.
ILP algorithms take examples E of a concept (such as a protein family) together with background knowledge B (such as a definition of molecular dynamics) and construct a hypothesis H which explains E in terms of B. For example, in protein fold domains, E might consist of descriptions of molecules separated into positive and negative examples of a particular fold (overall protein shape). This is exemplified in the diagram below for the fold ``4-helical-up-and-down-bundle''.
| Positive | Negative |
|---|---|
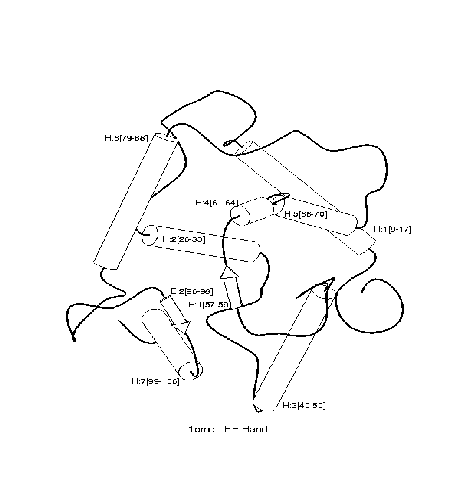
|
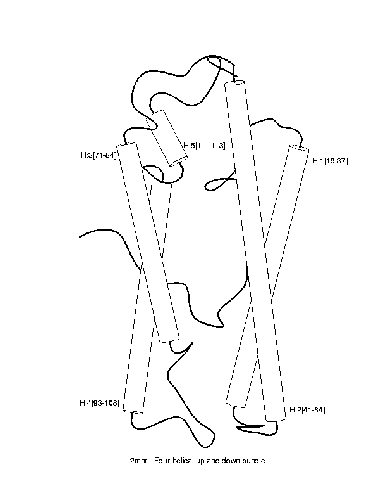
|
A possible hypothesis H describing this class of proteins is:
fold('Four-helical up-and-down bundle',P) :-
helix(P,H1),
length(H1,hi),
position(P,H1,Pos),
interval(1 =< Pos =< 3),
adjacent(P,H1,H2),
helix(P,H2).
The hypothesis is a definite clause consisting of a head (fold(..,..)) and a body (the conjunction length(..), .. helix(..)). In this case ``fold'' is the predicate involved in the examples and hypothesis, while ``length'', ``position'', etc. are defined by the background knowledge. A logic program is simply a set of such definite clauses. Each of E, B and H are logic programs.
In the context of knowledge discovery a distinct advantage of ILP over black box techniques, such as neural networks, is that a hypotheses such as that shown above can, in a straightforward manner, be automatically translated into the following piece of English text.
The protein P has fold class ``Four-helical up-and-down bundle'' if it contains a long helix H1 at a secondary structure position between 1 and 3, and H1 is followed by a second helix H2.Such explicit hypotheses can be used within the familiar human scientific discovery cycle of debate, criticism and refutation.
Discovery of biological function
Biological functions are regulated by the docking of small molecules (ligands) with sites on large molecules (proteins). Drugs, such as beta-blockers, mimic natural small molecules, such as adrenaline. Effectiveness of drugs depends on the correct shape and charge distribution of ligands. Thus beta-blockers block the binding of adrenaline, and so stop over-stimulation of heart muscle in patients prone to heart attacks.Results on scientific discovery applications of ILP are separated below between those related to small molecules (such as ligands) and those related to proteins.
Small molecules
Structure-activity prediction
The majority of pharmaceutical R&D is based on finding slightly improved variants of patented active drugs. This involves laboratories of chemists synthesising and testing hundreds of compounds almost at random. The average cost of developing a single new drug is around $300 million. In [3] it was shown that ILP system Golem [6] was capable of constructing rules which accurately predict the activity of untried drugs. Rules were constructed from examples of drugs with known medicinal activity. The accuracy of the rules was found to be slightly higher than traditional statistical methods. More importantly the easily understandable rules provided insights which were directly comparable to the relevant literature concerning the binding site of dihydrofolate reductase.Mutagenesis
In [4],[10] ILP system Progol [5] was used to predict the mutagenicity of chemical compounds taken from a previous study in which linear regression had been applied. Progol's predictive accuracy was equivalent to regression on the main set of 188 compounds and significantly higher (85.7% as opposed to 66.7%) on 44 compounds which had been discarded by the previous authors as unpredictable using regression. Progol's single clause solution for the 44 compounds was judged by the domain experts to be a new structural alert for mutagenesis.Pharmacophores
In a series of ``blind tests'' in collaboration with the pharmaceutical company Pfizer UK, Progol was shown [2] capable of re-discovering a 3D description of the binding sites (or pharmacophores) of ACE inhibitors (a hypertension drug) and an HIV-protease inhibitor (an anti-AIDS drug).Carcinogenicity
Last year Progol was entered into a world-wide carcinogenicity prediction competition run by the National Toxicology Program (NTP) in the USA. Progol was trained on around 300 available compounds, and made use of its earlier rules relating to mutagenicity. In the first round of the competition Progol produced the highest predictive accuracy of any automatic system entered [9].
| Method | Type | Accuracy | P |
|---|---|---|---|
| Ashby* | Chemist | 0.77 | 0.29 |
| Progol | ILP | 0.72 | 1.00 |
| RASH* | Biological potency analysis | 0.72 | 0.39 |
| TIPT* | Propositional ML | 0.67 | 0.11 |
| Bakale | Chemical reactivity analysis | 0.63 | 0.09 |
| Benigni | Expert-guided regression | 0.62 | 0.02 |
| DEREK | Expert system | 0.57 | 0.02 |
| TOPKAT | Statistical discrimination | 0.54 | 0.03 |
| CASE | Statistical correlation analysis | 0.54 | < 0.01 |
| COMPACT | Molecular modelling | 0.54 | 0.01 |
| Default | Majority class | 0.51 | 0.01 |
Proteins
Protein secondary structure prediction.
In [7] Golem was applied to one of the hardest open problems in molecular biology. The problem is as follows: given a sequence of amino acid residues, predict the placement of the main three dimensional sub-structures of the protein. The problem is of great interest to pharmaceutical companies involved with drug design. For this reason, over the last 20 years many attempts have been made to apply methods ranging from statistical regression to decision tree and neural net learning to this problem. Published accuracy results for the general prediction problem have ranged between 50 and 60%, very close to majority-class prediction rates. In our investigation we found the ability to make use of background knowledge from molecular biology, together with the ability to describe structural relations boosted the predictivity for a restricted sub-problem to around 80% on an independently chosen test set.Discovery of fold descriptions
Protein shape is usually described at various levels of abstraction. At the lower levels each family of proteins contains members with high sequence similarity. At the most abstract level folds describe proteins which have similar overall shape but are very different at the sequence level. The lack of understanding of shape determination has made protein fold prediction particularly hard. However, it is intriguing that although there are around 300 known folds, around half of all known proteins are members of the 20 most populated folds. In [12] Progol was applied to discover rules governing these 20 most populated protein folds. Average in class cross-validated prediction was around 70% and many of the rules were judged to be good characterisations of the fold classes by Michael Sternberg, a world-class protein prediction expert at the Imperial Cancer Research Fund in London.Conclusion
In his statement of the importance of this line of research to the Royal Society [11] Sternberg emphasised the aspect of joint human-computer collaboration in scientific discoveries. Science is an activity of human societies. It is our belief that computer-based scientific discovery must support strong integration into existing the social environment of human scientific communities. The discovered knowledge must add to and build on existing science. We believe that the ability to incorporate background knowledge and re-use learned knowledge together with the comprehensibility of the hypotheses, have marked out ILP as a particularly effective approach for scientific knowledge discovery.
Bibliography
-
- 1
-
I. Bratko and S. Muggleton.
Applications of inductive logic programming.
Communications of the ACM, 38(11):65-70, 1995. - 2
-
P. Finn, S. Muggleton, D. Page, and A. Srinivasan.
Pharmacophore discovery using the inductive logic programming system Progol.
Machine Learning, 30:241-271, 1998. - 3
-
R. King, S. Muggleton, R. Lewis, and M. Sternberg.
Drug design by machine learning: The use of inductive logic programming to model the structure-activity relationships of trimethoprim analogues binding to dihydrofolate reductase.
Proceedings of the National Academy of Sciences, 89(23):11322-11326, 1992. - 4
-
R. King, S. Muggleton, A. Srinivasan, and M. Sternberg.
Structure-activity relationships derived by machine learning: the use of atoms and their bond connectives to predict mutagenicity by inductive logic programming.
Proceedings of the National Academy of Sciences, 93:438-442, 1996. - 5
-
S. Muggleton.
Inverse entailment and Progol.
New Generation Computing, 13:245-286, 1995. - 6
-
S. Muggleton and C. Feng.
Efficient induction of logic programs.
In S. Muggleton, editor, Inductive Logic Programming, pages 281-298. Academic Press, London, 1992. - 7
-
S. Muggleton, R. King, and M. Sternberg.
Protein secondary structure prediction using logic-based machine learning.
Protein Engineering, 5(7):647-657, 1992. - 8
-
S. Muggleton and L. De Raedt.
Inductive logic programming: Theory and methods.
Journal of Logic Programming, 19,20:629-679, 1994. - 9
-
A. Srinivasan, , R.D. King S.H. Muggleton, and M. Sternberg.
Carcinogenesis predictions using ILP.
In N. Lavrac and S. Dzeroski, editors, Proceedings of the Seventh International Workshop on Inductive Logic Programming, pages 273-287. Springer-Verlag, Berlin, 1997.
LNAI 1297. - 10
-
A. Srinivasan, S. Muggleton, R. King, and M. Sternberg.
Theories for mutagenicity: a study of first-order and feature based induction.
Artificial Intelligence, 85(1,2):277-299, 1996. - 11
-
M. Sternberg, R. King, R. Lewis, and S. Muggleton.
Application of machine learning to structural molecular biology.
Philosophical Transactions of the Royal Society B, 344:365-371, 1994. - 12
-
M. Turcotte, S.H. Muggleton, and M.J.E. Sternberg.
Protein fold recognition.
In C.D. Page, editor, Proc. of the 8th International Workshop on Inductive Logic Programming (ILP-98), LNAI 1446, pages 53-64, Berlin, 1998. Springer-Verlag.